



Une mise en place optimisée des essais cliniques
Afin de permettre la mise en place rapide d’un essai sur un vaccin prometteur tout en s’assurant de la qualité du produit et de la sécurité des participants, les autorités sanitaires européennes ont mis en place des procédures dites “accélérées” pour l’évaluation des demandes d’essais cliniques.En France, un essai clinique ne peut démarrer sans avoir reçu au préalable un avis favorable d’un comité de protection des personnes (CPP) et une autorisation de l’ANSM.
Pour permettre une préparation plus efficace des demandes d’essais cliniques, les autorités sanitaires échangent régulièrement avec les promoteurs (institution, organisme ou laboratoire pharmaceutique responsable de l’essai clinique), afin que les dossiers déposés prennent directement en compte l’ensemble des exigences de qualité et de sécurité pour les patients. La procédure d’évaluation des demandes d’essai clinique est ainsi facilitée.
À retenir
Au 1er juillet 2022, 6 vaccins ont passé l’étape des essais cliniques et obtenu une autorisation de mise sur marché de l’Agence européenne des médicaments (EMA) :
Différents candidats vaccins sont par ailleurs toujours à l’étude à travers le monde. Certains exploitent d’autres plateformes vaccinales comme le virus entier inactivé ou atténué, d’autres sont des adaptations des vaccins déjà autorisés, modifiés au regard des nouveaux variants.
- Comirnaty (Pfizer) et Spikevax (Moderna) ; ARN messager
- Jcovden (Johnson & Johnson) et Vaxzevria (AstraZeneca) ; vecteur viral non réplicatif
- Nuvaxovid (Novavax) ; protéine sous-unitaire
- Valneva ; inactivé
Différents candidats vaccins sont par ailleurs toujours à l’étude à travers le monde. Certains exploitent d’autres plateformes vaccinales comme le virus entier inactivé ou atténué, d’autres sont des adaptations des vaccins déjà autorisés, modifiés au regard des nouveaux variants.
Une surveillance nationale et internationale des essais cliniques
L’ANSM assure, en lien avec les promoteurs des essais cliniques, l’examen des évènements indésirables graves survenus pendant toute la durée d’un essai mené chez des volontaires sains.En cas de risque pour la sécurité des participants, le promoteur ou l’ANSM peut décider à tout moment de suspendre l’essai clinique afin d’en réévaluer le rapport bénéfice/risque, puis d’en modifier la conduite si nécessaire.
Une coopération internationale est également mise en place afin d’être attentif aux signaux qui pourraient survenir dans d’autres pays où sont évalués les vaccins, pour que des mesures appropriées soient prises sans délai.
Une évaluation en temps réel des vaccins avant AMM : la "rolling review"
L’évaluation des demandes d’autorisation de mise sur le marché des vaccins contre le Covid-19 est réalisée en continu, c’est-à-dire à partir des données transmises en temps réel par les fabricants.Habituellement, les données sur l’efficacité, la sécurité et la qualité d’un médicament, ainsi que tous les documents requis pour obtenir une AMM, sont soumis en une seule fois avant leur évaluation dans une demande officielle déposée par le fabricant. Dans le cas de l’examen continu mis en place par l’Agence européenne du médicament, nommé « rolling review » en anglais, les agences européennes analysent les données au fur et à mesure de leur collecte pendant les études en cours. La période d’évaluation est ainsi écourtée tout en garantissant le même niveau de sécurité pour les personnes :
- L’évaluation commence dès que les premières données sur le vaccin sont disponibles. En général, elles proviennent d’abord des études en laboratoire (données non cliniques) ;
- Cela ne signifie pas que l’on peut conclure rapidement sur l’innocuité et l’efficacité du vaccin. Seule l’évaluation des données issues des études cliniques portant sur un grand nombre de volontaires permettront de s’en assurer.
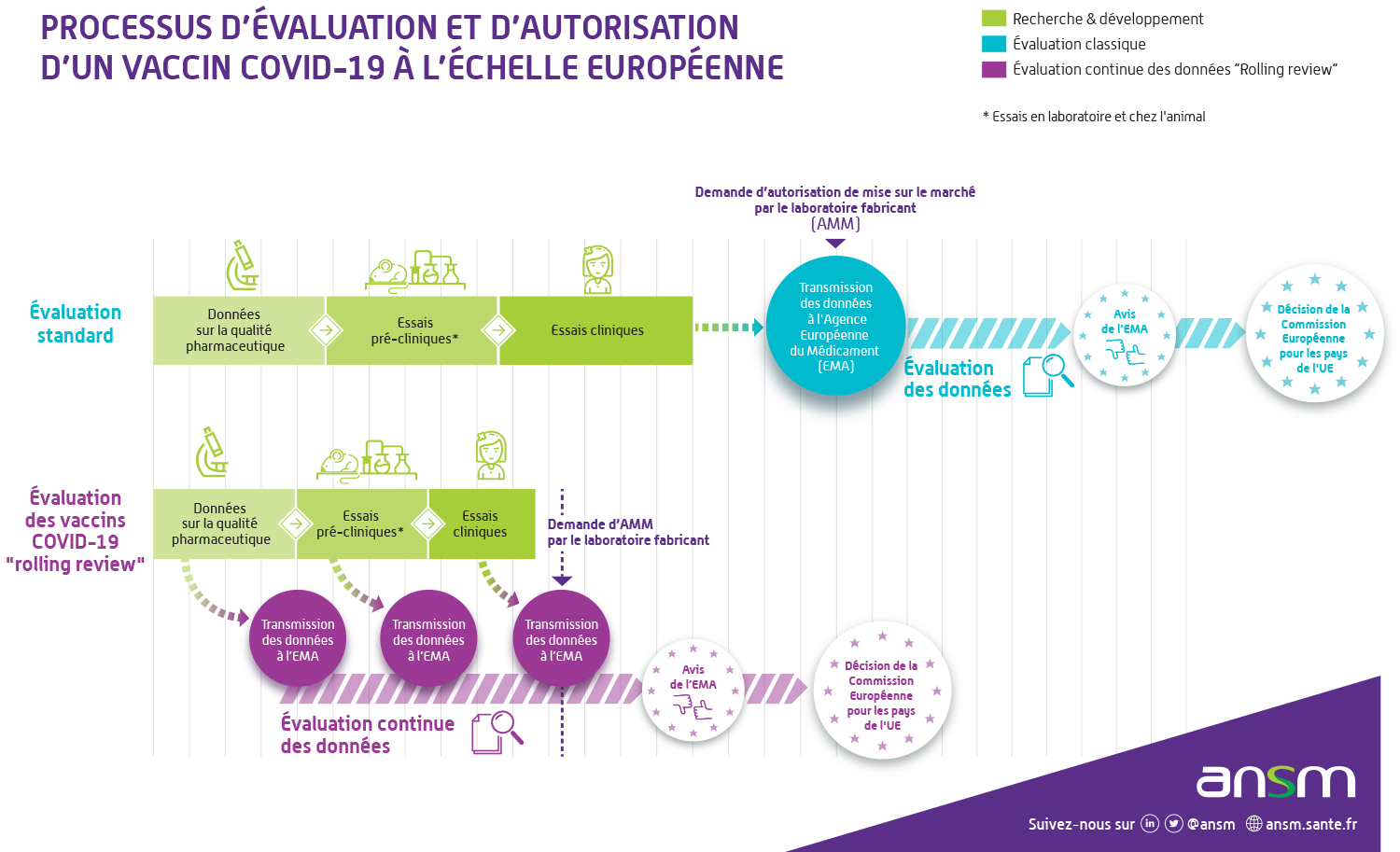
En France, c’est une task force au sein de l’ANSM qui contribue à l’examen de ces données. Elle est également mobilisée de façon réactive pour prendre en charge l’instruction des essais cliniques de vaccins contre le Covid-19 (phase III principalement) en articulation avec les comités de protection des personnes (CPP).
Vaccins en cours d’évaluation continue : consultez le site de l’EMA
Vaccins en cours d’évaluation continue
Des autorisations de mise sur le marché conditionnelles
Les autorisations de mise sur le marché (AMM) sont délivrées par la Commission européenne à l’issue de cette évaluation en rolling review, et sont valables dans tous les Etats membres de l’Union européenne. Face à l’urgence de santé publique dans un contexte exceptionnel de pandémie, les AMM des vaccins contre le Covid-19 sont dites “conditionnelles”. Ce n’est toutefois pas la première fois que des AMM conditionnelles sont délivrées. Auparavant, ce type d’autorisation a déjà été utilisé pour des traitements de maladies rares par exemple.Une AMM conditionnelle permet en effet l’autorisation de médicaments répondant à un besoin médical non satisfait avant que des données à long terme sur l'efficacité et la sécurité ne soient disponibles. Cela est possible uniquement si les bénéfices de la disponibilité immédiate du médicament sont plus grands que le risque inhérent au fait que toutes les données ne sont pas encore disponibles. L’AMM conditionnelle implique strictement les mêmes contrôles qu’une autorisation de mise sur le marché standard pour garantir un niveau élevé de sécurité aux patients.
Une fois qu'une AMM conditionnelle a été accordée, les laboratoires doivent fournir les données complémentaires provenant d'études nouvelles ou en cours dans des délais fixés par l’EMA pour confirmer la balance bénéfice/risque positive.
Une AMM conditionnelle est accordée pour un an et peut être renouvelée. Lorsque les autorités européennes ont reçu et évalué toutes les données complémentaires exigées, l’AMM conditionnelle peut être convertie en une AMM standard.
